
If you were to look at the table of contents for the world’s leading computational biology journals over the past week, one theme overwhelmingly dominates the conversation: Spatial Transcriptomics (ST) and Biological Foundation Models.
Historically, single-cell RNA sequencing (scRNA-seq) was the gold standard for understanding cellular heterogeneity. However, scRNA-seq requires tissue dissociation—it gives researchers a high-resolution list of the "ingredients" in a biological sample, but it completely destroys the structural "recipe." We knew what cells were present, but we lost all information regarding where they were located and who they were interacting with.
Spatial Transcriptomics solved this by preserving the 2D (and now 3D) coordinates of gene expression. But it introduced a massive computational bottleneck: how do you integrate millions of spatial coordinates, highly sparse gene expression matrices, and billions of intercellular interaction networks without requiring a supercomputer?
In the first week of July 2026, the journal Bioinformatics (Oxford Academic) published a triad of revolutionary papers that have effectively solved this computational bottleneck. These papers introduce structural-guided fusion algorithms, automated cross-dataset harmonizations, and—most importantly—a framework that allows billion-parameter biological AI models to run on standard consumer-grade laptops.
For doctoral scholars and early-career researchers, these publications represent a massive paradigm shift. This 3,000-word deep dive explores these three groundbreaking papers, breaks down the computational mechanics driving them, and outlines how you can leverage these discoveries for your own high-impact research.
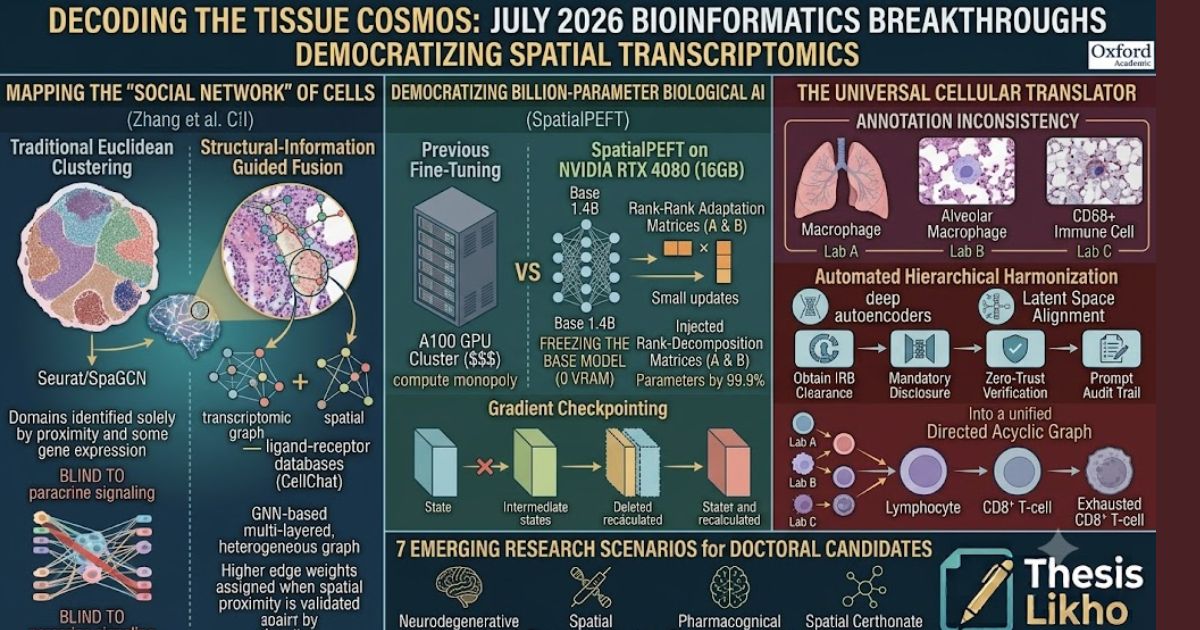
Breakthrough 1: Mapping the "Social Network" of Cells
The Paper: Structural-information Guided Fusion for spatial domain identification from Spatial Transcriptomics (Zhang et al., July 2026, Bioinformatics).
The Core Problem: The Blind Spot of Early ST Models
When analyzing Spatial Transcriptomics data, the primary goal is "spatial domain identification"—grouping cells into functional zones (e.g., identifying the necrotic core of a tumor versus the invasive leading edge).
Until last week, most ST clustering algorithms (like Seurat or SpaGCN) relied almost exclusively on two metrics:
- Gene Expression Profiles: Which genes are turned on/off?
- Euclidean Spatial Coordinates: Is Cell A physically next to Cell B?
However, this approach suffers from a critical biological blind spot. Just because two cells are physically adjacent does not mean they are biologically interacting. Conversely, cells that are slightly further apart might be communicating intensely via paracrine signaling (secreting cytokines or chemokines). Existing algorithms were essentially mapping a city by looking at the physical addresses of houses, completely ignoring the phone calls and emails being sent between them.
The Solution: Structural-Information Guided Fusion
Zhang et al. (July 2026) introduced a novel algorithmic framework that fuses Graph Neural Networks (GNNs) with deep structural biological information. Instead of just creating a simple spatial graph (where edges connect nearest physical neighbors), their model constructs a multi-layered, heterogeneous graph.
The Computational Mechanism
- Transcriptomic Graph: The model calculates the Pearson correlation coefficient between the gene expression vectors of different spots. If two spots share highly similar transcriptomic signatures, they are connected, regardless of spatial distance.
- Spatial Graph: A standard K-Nearest Neighbor (KNN) graph is built using the physical (x, y) coordinates of the tissue slice.
- The Interaction Layer (The Breakthrough): The algorithm integrates known ligand-receptor (L-R) interaction databases (like CellChat or NicheNet). It actively searches for physical proximity combined with the simultaneous expression of a ligand in Cell A and its corresponding receptor in Cell B.
- Guided Fusion: Using an attention mechanism, the deep learning model fuses these three graphs. The model learns to assign higher weights to edges where spatial proximity is validated by active ligand-receptor signaling.
Research Significance for Scholars
This paper fundamentally changes how we study the Tumor Microenvironment (TME). By utilizing this structural-guided fusion, researchers can now accurately map how Cancer-Associated Fibroblasts (CAFs) actively suppress CD8+ T-cells in 3D space.
For a PhD student, applying this fusion architecture to novel datasets (such as Alzheimer's brain tissue to map microglia-neuron interactions) is a guaranteed pathway to a Q1 SCI-indexed publication. The code for these GNN architectures is becoming the new standard for computational oncology.
Breakthrough 2: Democratizing Billion-Parameter AI in Biology
The Paper: SpatialPEFT: A unified parameter-efficient fine-tuning framework for large spatial transcriptomics foundation models (July 2026, Bioinformatics).
The Core Problem: The Compute Monopoly
In 2024 and 2025, the bioinformatics world saw the rise of "Biological Foundation Models"—massive AI networks like scGPT, Geneformer, and UCE (Universal Cell Embedding). These models, trained on tens of millions of single cells across hundreds of tissues, act like the ChatGPT of biology. They have a deep, pre-trained understanding of fundamental gene networks.
To use these models for a specific project (e.g., analyzing pediatric leukemia), researchers need to "fine-tune" them on their own specific datasets. The problem? These models contain up to 1.4 billion parameters. Fine-tuning them required clusters of A100 GPUs, hardware that costs hundreds of thousands of dollars. This created a "compute monopoly," where only elite, massively funded institutions (like the Broad Institute or Sanger Institute) could participate in cutting-edge AI biology.
The Solution: SpatialPEFT and LoRA
Published just days ago, the SpatialPEFT framework shatters this monopoly. The researchers successfully adapted advanced NLP (Natural Language Processing) techniques to biological data, allowing a 1.4-billion-parameter foundation model to be fine-tuned on a single 16 GB consumer-grade GPU (like an NVIDIA RTX 4080 found in a standard gaming laptop).
The Computational Mechanism: Low-Rank Adaptation (LoRA)
To understand why this is revolutionary, you must understand the math behind LoRA.
Normally, when you fine-tune a neural network, you must update every single weight (parameter) in the model. Updating 1.4 billion weights requires massive VRAM to store the gradient calculations.
SpatialPEFT uses LoRA to bypass this:
- Freezing the Base Model: The algorithm "freezes" the 1.4 billion pre-trained weights. They are locked in place and require zero memory for gradient updates.
- Injecting Rank-Decomposition Matrices: The algorithm injects two tiny, trainable matrices ($A$ and $B$) alongside the frozen layers. Instead of updating a massive $10,000 \times 10,000$ weight matrix, LoRA only updates a $10,000 \times 8$ matrix ($A$) and an $8 \times 10,000$ matrix ($B$).
- Gradient Checkpointing: Furthermore, SpatialPEFT utilizes gradient checkpointing, which strategically deletes intermediate memory states during the forward pass and re-calculates them on the fly during the backward pass, drastically reducing memory spikes.
By taking the product of these small matrices ($A \times B$), the model approximates the massive weight updates. This reduces the trainable parameters by 99.9%, while maintaining 99% of the model's predictive accuracy.
Research Significance for Scholars
This is arguably the most important paper of the year for independent researchers and universities in developing nations. SpatialPEFT democratizes biological AI.
A master's or PhD student can now download a massive, pre-trained spatial foundation model, load SpatialPEFT on their laboratory PC, and fine-tune it to their highly specific niche dataset (e.g., the spatial transcriptomics of drought-stressed rice roots) in a matter of hours. This opens up thousands of new, highly publishable thesis topics that were previously blocked by hardware limitations.
Breakthrough 3: The Universal Cellular Translator
The Paper: Cross-dataset annotation harmonization for cell-type hierarchy construction (Zhou et al., July 2026, Bioinformatics).
The Core Problem: The Tower of Babel in Single-Cell Biology
As researchers across the globe sequence different tissues, they upload their data to public repositories like the NCBI Gene Expression Omnibus (GEO). We currently have exabytes of spatial and single-cell data freely available. However, integrating this data is a nightmare due to annotation inconsistency.
Imagine three different research labs sequencing the human lung:
- Lab A annotates a specific cell as a "Macrophage."
- Lab B annotates the exact same cell type as an "Alveolar Macrophage."
- Lab C annotates it as an "Immune Cell (CD68+)."
When a computational biologist tries to merge these three datasets to train a machine learning model, the computer treats these as three entirely different entities. This lack of a standardized cell-type hierarchy has hindered the creation of a true "Human Cell Atlas."
The Solution: Automated Hierarchical Harmonization
Zhou et al. (July 2026) introduced a sophisticated computational framework that acts as a universal translator for biological datasets. Instead of forcing all datasets into a flat, rigid naming convention, the algorithm builds a Directed Acyclic Graph (DAG)—a hierarchical tree of cell types.
The Computational Mechanism
- Latent Space Alignment: The algorithm projects the gene expression profiles of all cells from diverse datasets into a shared mathematical "latent space" using deep autoencoders.
- Semantic Similarity Scoring: The model calculates the transcriptomic distance between the clusters. It recognizes that Lab B's "Alveolar Macrophage" is a highly specific sub-cluster perfectly nested within Lab A's broader "Macrophage" cluster.
- Bottom-Up Propagation: The algorithm automatically maps these relationships, constructing a unified, multi-resolution hierarchy. It links specific terminal states (e.g., "Exhausted CD8+ T-cell") up to their parent nodes ("CD8+ T-cell"), and further up to root nodes ("Lymphocyte").
Research Significance for Scholars
For bioinformaticians, data cleaning and harmonization usually consume 70% of a project's timeline. This algorithmic harmonization tool drastically reduces that burden.
If your thesis involves conducting a "meta-analysis" of a specific disease—say, pulling down 20 different spatial transcriptomics datasets of triple-negative breast cancer from GEO to find universal biomarkers—this tool is indispensable. It allows you to merge millions of cells seamlessly, increasing your statistical power and paving the way for high-impact publications in journals like Nature Methods or Cell Systems.
Synthesizing the Big Picture: What Does This Mean for the Future?
If we look at these three July 2026 papers holistically, we see a clear trajectory for the future of bioinformatics:
- Biology is now fundamentally a Data Science. The days of simple differential expression analysis (finding which genes are "up" or "down") are over. We are now modeling tissues as complex, dynamic, interactive networks.
- Accessibility is the new Innovation. The shift from raw computational power to algorithmic efficiency (like LoRA and SpatialPEFT) means that the next great breakthrough might not come from a massive pharmaceutical company, but from a graduate student working on a laptop in a university lab.
- Context is Everything. Whether it is using GNNs to map intercellular structural interactions or building hierarchical DAGs for cell annotations, bioinformatics is moving away from isolated data points and toward deep contextual integration.
7 Emerging Research Scenarios for Doctoral Candidates in 2026
If you are a master’s student or PhD candidate looking to leverage these July 2026 breakthroughs for your dissertation, here are seven highly actionable, publication-ready research avenues:
- Neurodegenerative Spatial Mapping: Utilize Structural-information Guided Fusion to map the spatial relationship between amyloid-beta plaques and reactive astrocyte states in Alzheimer's disease models.
- Agri-Genomics Foundation Tuning: Apply SpatialPEFT to fine-tune a mammalian foundation model using plant spatial transcriptomics data (e.g., Arabidopsis leaf cross-sections) to identify novel drought-resistance pathways.
- Autoimmune Cross-Tissue Analysis: Use Zhou et al.’s Harmonization algorithm to integrate single-cell datasets from Rheumatoid Arthritis (joint fluid) and Lupus (kidney tissue) to find universal, pan-autoimmune exhausted T-cell signatures.
- Spatial Pharmacogenomics: Map how the spatial architecture of a solid tumor changes before and after the administration of immunotherapy, using GNNs to track the physical infiltration of immune cells.
- Viral Tropism and Tissue Architecture: Analyze spatial transcriptomics from COVID-19 or Influenza-infected lung tissues to map exactly which cellular neighborhoods are most vulnerable to viral entry based on receptor co-expression.
- Embryonic Developmental Trajectories: Apply hierarchical DAG harmonization to spatial data of developing embryos, tracing how a single stem cell lineage branches into complex, spatially distinct organ systems over time.
- Benchmarking Parameter Efficiency: Conduct a purely computational study comparing the memory usage, training time, and biological accuracy of LoRA versus standard fine-tuning across different biological foundation models.
Elevating Your Bioinformatics Research with Thesislikho.com
The bioinformatics breakthroughs of July 2026 are thrilling, but implementing them is incredibly daunting. Downloading a 1.4-billion-parameter foundation model, setting up a Python environment with PyTorch and CUDA dependencies, writing the scripts for Low-Rank Adaptation, and executing a Graph Neural Network requires a level of computer science expertise that many biology-focused researchers simply do not have yet.
The transition from a brilliant biological hypothesis to a published, mathematically sound computational pipeline is where many scholars stall. This is exactly where Thesislikho.com steps in as your specialized academic partner.
We do not just proofread; we provide the deep, technical scaffolding required to execute world-class bioinformatics research.
How Thesislikho Elevates Your Computational Research:
- Code Debugging and Pipeline Development: Are you struggling to get SpatialPEFT running on your local machine? Are your PyTorch tensors throwing dimensionality errors when you try to fuse your spatial and transcriptomic graphs? Our team of expert bioinformaticians and data scientists provides direct coding consultancy. We help you troubleshoot Python and R scripts, optimize your algorithms for memory efficiency, and ensure your code compiles flawlessly.
- Methodological Rigor for SCI Journals: When publishing in top-tier computational journals like Bioinformatics or Briefings in Bioinformatics, your methodology chapter must be mathematically rigorous. We assist you in documenting your algorithmic architecture—explaining your loss functions, your attention mechanisms, and your graph edge constructions—in the precise, formal language demanded by peer reviewers.
- Data Visualization and Formatting: Spatial transcriptomics generates stunning visual data, but formatting these spatial plots, UMAPs, and hierarchical trees to meet the high-resolution, specific dimensions of Oxford Academic or Nature Publishing Group is tedious. Our technical formatting team ensures your figures are publication-ready.
- Navigating Cross-Disciplinary Literature: Bioinformatics sits at the intersection of computer science, statistics, and molecular biology. If you are struggling to synthesize a literature review that bridges the mathematics of LoRA with the biology of the tumor microenvironment, our academic consultants help you weave these disparate fields into a cohesive, compelling narrative.
- PhD Proposal Development: If you want to base your doctoral research on these new spatial foundation models, we help you draft a highly competitive PhD synopsis. We ensure your research question highlights a clear, novel gap in the literature, positioning you as an ideal candidate for admission into premier computational biology programs.
The bioinformatics landscape has fundamentally shifted as of July 2026. The tools required to unlock the spatial secrets of the cell are now openly available and computationally efficient. Do not let coding errors, mathematical formatting, or structural writing challenges prevent you from contributing to this biological revolution.
Partner with the technical and academic experts at Thesislikho.com to ensure your computational research is rigorous, reproducible, and published at the absolute highest levels of the scientific community.
As spatial foundation models become easier to fine-tune on consumer hardware, how do you see this altering the balance of scientific power between massive, well-funded institutions and independent academic researchers in the coming decade?
Call / WhatsApp: +91 96438 02216
Website:www.ThesisLikho.com